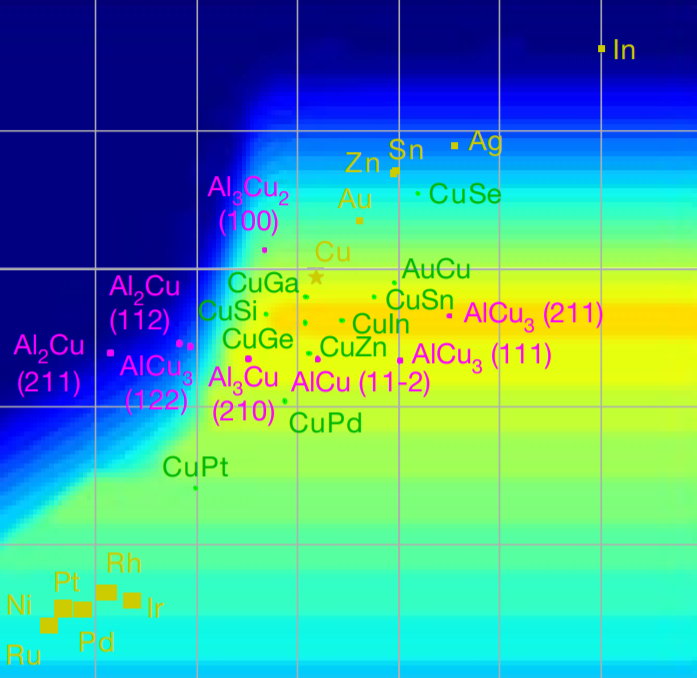
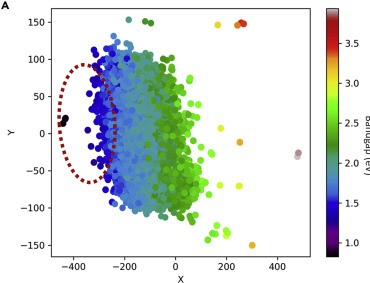
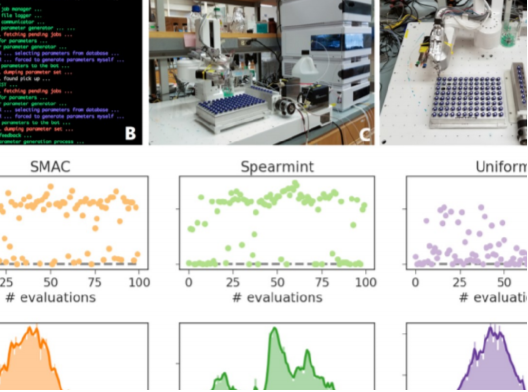
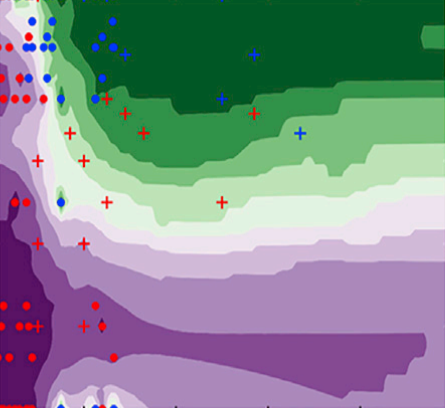
The rapid increase in global energy demand and the need to replace carbon dioxide (CO2)-emitting fossil fuels with renewable sources have driven interest in chemical storage of intermittent solar and wind energy. Particularly attractive is the electrochemical reduction of CO2 to chemical feedstocks, which uses both CO2 and renewable energy. Copper has been the predominant electrocatalyst for this reaction when aiming for more valuable multi-carbon products, and process improvements have been particularly notable when targeting ethylene. However, the energy efficiency and productivity (current density) achieved so far still fall below the values required to produce ethylene at cost-competitive prices. Here we describe Cu-Al electrocatalysts, identified using density functional theory calculations in combination with active machine learning, that efficiently reduce CO2 to ethylene with the highest Faradaic efficiency reported so far. This Faradaic efficiency of over 80 per cent (compared to about 66 per cent for pure Cu) is achieved at a current density of 400 milliamperes per square centimetre (at 1.5 volts versus a reversible hydrogen electrode) and a cathodic-side (half-cell) ethylene power conversion efficiency of 55 ± 2 per cent at 150 milliamperes per square centimetre. We perform computational studies that suggest that the Cu-Al alloys provide multiple sites and surface orientations with near-optimal CO binding for both efficient and selective CO2 reduction. Furthermore, in situ X-ray absorption measurements reveal that Cu and Al enable a favourable Cu coordination environment that enhances C–C dimerization. These findings illustrate the value of computation and machine learning in guiding the experimental exploration of multi-metallic systems that go beyond the limitations of conventional single-metal electrocatalysts.